 400-166-8600400-166-8600
400-166-8600400-166-8600
帕金森病(Parkinson’s disease,PD)
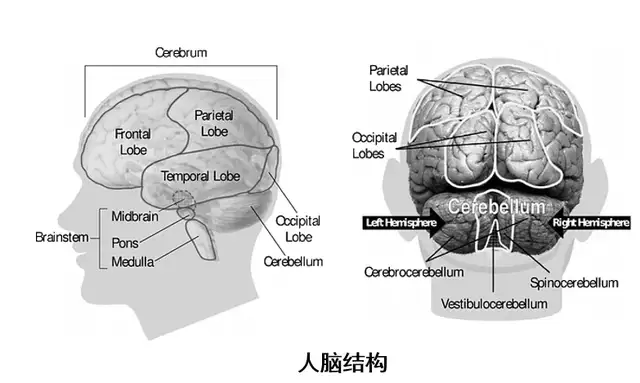
是一種常見的神經系統變性疾病,老年人多見,平均發病年齡為60歲左右,40歲以下起病的青年帕金森病較少見。我國65歲以上人群PD的患病率大約是1.7%。
大部分帕金森病患者為散發病例,僅有不到10%的患者有家族史。帕金森病最主要的病理改變是中腦黑質多巴胺(dopamine, DA)能神經元的變性死亡,由此而引起紋狀體DA含量顯著性減少而致病。
導致這一病理改變的確切病因仍不清楚,遺傳因素、環境因素、年齡老化、氧化應激等均可能參與PD多巴胺能神經元的變性死亡過程。
病因
有研究表明,黑質(substantia nigra, SN)中多巴胺能神經元喪失會導致患者表現出的運動功能障礙。
PD致病因素多種多樣,但有不少證據表明,線粒體功能缺陷是很重要的一項。例如:編碼維持線粒體質量控制蛋白的PARK7、PARK6和PARK2基因突變能引起早發型PD。
由于多巴胺能神經元對代謝有很高的需求,因此造成其對線粒體功能障礙十分敏感。而線粒體氧化磷酸化(OXPHOS)的持續刺激,是以線粒體氧化損傷增加為代價的。
研究表明,PD患者SN中mtDNA完整性的喪失與功能性線粒體復合物I(MCI)的喪失存在一定的相關性。然而,這種MCI獲得性損傷究竟是PD疾病進程中的一種副產品還是疾病的驅動因素還不得而知。
最新研究
為了探究線粒體-黑質-帕金森之間的關系,美國西北大學的研究團隊進行了研究,最新結果發表在《Nature》上,論文標題為:“Disruption of mitochondrial complex I induces progressive parkinsonism"。

本文關鍵詞
帕金森病(Parkinson’s disease,PD)
黑質(substantia nigra, SN)
多巴胺能神經元
功能性線粒體復合物I(MCI)
NDUFS4亞基
線粒體氧化磷酸化(OXPHOS)
濃縮就是精華版
使用交叉遺傳學來破壞小鼠多巴胺能神經元的MCI功能。MCI的破壞誘導了代謝的warburg樣改變,使神經元存活,但引發運動學習和精細運動缺陷。
因此,單是MCI功能障礙就足以引起進行性的類人帕金森病,在這種情況下,黑素多巴胺釋放的缺失對運動功能障礙起關鍵作用。
并且人員還探究了不同類型的運動功能損傷,與不同部位多巴胺釋放的相關性。
這些研究挑戰了該疾病長期以來的主流的觀點。
細胞研究離不開血清的支持,
點擊下方小程序或結尾的“閱讀原文"
了解更多血清產品
Ausbian進口特級胎牛血清
小程序
結果1:
采用Cre-loxP構建Ndufs2缺失小鼠品系。
得出最終結論:MCI的喪失會引起代謝重編程
大約在出生后第20天(P20)),條件下ndufs2敲除小鼠(cNdufs2?/?)的外觀和大運動行為正常(MCI蛋白通常有20-40天的生命周期14,因此可能在出生后早期沒有明顯表型)。
在P20和P30之間,SN多巴胺能神經元中的線粒體成為三磷酸腺苷(ATP)的凈消費者,而不是生產者,且線粒體嵴變化明顯[這種變化在線粒體內膜(IMM)電位的敏感性中很明顯——使用電位染料四甲基羅丹明(tetramethylrhodamine)對腺嘌呤核苷酸轉運體(ANT)阻斷的敏感性測量]。
cNdufs2?/?多巴胺能神經元中的體細胞線粒體密度是正常的。但是在許多cNdufs2?/?多巴胺能神經元中,線粒體嵴的結構發生了改變與電子傳遞鏈基因的下調一致。
分離野生型和cNdufs2?/?SN多巴胺能神經元的mRNA,并對其進行測序。在cNdufs2?/?多巴胺能神經元中存在大量的代謝重編程——一種類似warburg的效應。
促進糖酵解的基因編碼蛋白上調,與OXPHOS相關的基因下調。編碼糖酵解抑制因子的基因也被下調。
野生型多巴胺能神經元,寡霉素抑制線粒體復合物 V 導致 ATP 水平急劇下降。與之相比,在 cNdufs2?/? 多巴胺能神經元,該比率因糖酵解抑制而下降。

結果2:
除了觸發代謝重編程外,Ndufs2的缺失還會導致軸突生長和轉運相關基因(如Tubb3、Uchl1、Wnt5a、Sema3g、Nefl和Prkca)、突觸傳遞相關基因(如Syt1、Syt3、Syt17、Syn2和Scna)的表達發生重大變化,DA合成/儲存(如Th和Vamp2)和突觸前調節(如Drd2, Chrna4和Chrna6)。背側紋狀體中酪氨酸羥化酶(TH)蛋白水平在P30左右下調,與線粒體OXPHOS的喪失平行。
對紋狀體組織的液相色譜和質譜分析進一步驗證cNdufs2-/-小鼠紋狀體DA合成明顯下降,此外,有助于驅動起搏的環核苷酸門控陽離子通道電流也明顯減少。
結果3:
Ndufs2缺失后的體樹突轉移。
到P60,與多巴胺能信號相關的軸突蛋白的丟失由背側紋狀體擴大到腹側紋狀體。
cNdufs2-/-小鼠SN多巴胺能神經元胞體樹突區域中的酪氨酸羥化酶表達降低至對照組一半左右。
DA釋放量下降約75%。
組織學分析得出結論:在cNdufs2?/?小鼠病理進化的這個階段,多巴胺能信號傳遞標記的丟失反映了表型下調,而不是明顯的神經退行性變;也就是說,黑質紋狀體軸突的分支形態是否保留仍有待確定。
對多巴胺能神經元進行標記后,發現cNdufs2?/?SN多巴胺能神經元的體樹突遞質表型的下降與對外部興奮性刺激反應的損失并不緊密匹配。
結果4:
帕金森癥的出現。
與在整個基底神經節中DA迅速耗盡的傳統PD模型相比,cNdufs2-/-小鼠的病理分期能夠評估DA釋放的區域缺陷如何與行為相關聯。
當背側紋狀體DA釋放在P30左右下降到接近檢測閾值時,cNdufs2?/?小鼠失去了執行聯想學習任務的能力,而聯想學習任務被認為依賴于DA依賴的紋狀體突觸可塑性。
在P30的全體性左旋多巴治療(6 mg kg?1)可以恢復這一記憶任務,但在P60的后期治療卻不能。
在通過小鼠從前爪去除粘合劑所花費的時間來評估精細運動技能的實驗中,cNdufs2-/-小鼠完成任務時間明顯延長,同時也表現出較差的曠場探索行為表現。
P60的cNdufs2-/-小鼠僅表現出輕微的步態障礙,到了P100才會表現出后肢張開、爪子位置異常和步幅改變等特征。而在P120-150期間,大約有40%的SN多巴胺能神經元丟失。
結果5:
運動障礙的黑質決定簇。
分別向小鼠背側紋狀體或SN中立體定位注射攜帶AADC(可將左旋多巴轉化為DA)的AAV,以及隨后對小鼠曠場步態的分析,證明黑質多巴胺釋放喪失對于粗大運動缺陷而言是必要因素。
★ 總結
這項研究,不僅證明多巴胺能神經元中MCI功能喪失足以引發進行性的、軸突先行的功能喪失和左旋多巴反應性帕金森病,還證明背側紋狀體的DA耗竭對于聯想運動學習和精細動作而言是必要的,但黑質的DA釋放缺陷才會引起類似于臨床PD患者表現出的粗大運動損傷特征。
該模型不僅可以研究復合物 I 缺陷在疾病中的作用,還可以具有評估治療策略的潛力。
參考文獻:González-Rodríguez, Patricia et al. “Disruption of mitochondrial complex I induces progressive parkinsonism." Nature vol. 599,7886 (2021): 650-656.
doi:10.1038/s41586-021-04059-0
 當前位置:
當前位置: